
LIBMELDY 2-10 x 1 000 000 cellules - ml, dispersion pour perfusion, boîte métallique de 1 poche ( set perfusion) de 20 ml
Dernière révision : 26/08/2022
Taux de TVA : 0%
Laboratoire exploitant : PHARMA BLUE
Source :

Libmeldy est indiqué dans le
traitement de la leucodystrophie métachromatique (LDM) caractérisée par des
mutations bialléliques du gène de l'arylsulfatase A (ARSA) entraînant une réduction de
l'activité enzymatique de l'ARSA:
- chez les enfants atteints de la forme infantile tardive ou juvénile précoce, sans manifestations cliniques de la maladie,
- chez les enfants atteints de la forme juvénile précoce, présentant des manifestations cliniques précoces de la maladie, qui ont conservé la capacité de marcher indépendamment et avant l'apparition du déclin cognitif (voir rubrique Propriétés pharmacodynamiques).
- chez les enfants atteints de la forme infantile tardive ou juvénile précoce, sans manifestations cliniques de la maladie,
- chez les enfants atteints de la forme juvénile précoce, présentant des manifestations cliniques précoces de la maladie, qui ont conservé la capacité de marcher indépendamment et avant l'apparition du déclin cognitif (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Antécédent de traitement par thérapie génique à base de cellules souches hématopoïétiques.
Les contre-indications aux agents de mobilisation et de conditionnement myéloablatif doivent être prises en compte.
Traçabilité
Les exigences en matière de traçabilité des médicaments de thérapie cellulaire innovante doivent s'appliquer. Afin de garantir la traçabilité, le nom du produit, le numéro de lot et le nom du patient traité doivent être conservés pendant une durée de 30 ans.
Usage autologue
Libmeldy est destiné uniquement à un usage autologue et ne doit en aucun cas être administré à d'autres patients. Ne pas perfuser Libmeldy si les informations figurant sur les étiquettes du produit et sur la fiche d'information du lot ne correspondent pas à l'identité du patient.
Phase de progression rapide de la maladie
Le traitement par Libmeldy doit être réalisé avant que la maladie n'entre dans sa phase de progression rapide.
L'éligibilité au traitement par Libmeldy doit d'abord être évaluée par
le médecin traitant au moyen d'un examen neurologique complet, d'une
évaluation de la fonction motrice et d'une évaluation neurocognitive,
selon l'âge du patient.
Avant le début du prélèvement cellulaire, le médecin traitant doit
s'assurer que l'état clinique du patient ne s'est pas détérioré.
Ensuite, avant le début du conditionnement, le médecin traitant doit
s'assurer que l'administration d'une thérapie génique autologue par
CSPH reste cliniquement appropriée pour le patient et que le traitement
par Libmeldy reste indiqué.
Médicaments de mobilisation et conditionnement myéloablatif
Les mises en garde et les précautions d'emploi des médicaments de mobilisation et de conditionnement myéloablatif doivent être prises en compte.
Complications liées aux cathéters veineux centraux (CVC), notamment les infections et les thromboses veineuses
Des infections liées à l'utilisation de CVC ont été signalées dans des études cliniques et il existe un risque de thrombose associé au CVC. Les patients doivent faire l'objet d'une surveillance étroite pour détecter d'éventuelles infections et événements liés au cathéter.
Réactions liées à l'hypersensibilité et à la perfusion
Le diméthylsulfoxyde (DMSO), l'un des excipients de Libmeldy, est connu pour provoquer possiblement des réactions anaphylactiques après une administration parentérale. Les patients qui n'ont pas été exposés auparavant au DMSO doivent faire l'objet d'une surveillance étroite. Les signes vitaux (pression artérielle, rythme cardiaque et saturation en oxygène) et l'apparition de tout symptôme doivent être surveillés avant le début de la perfusion, environ toutes les dix minutes pendant la perfusion, puis toutes les heures, pendant 3 heures, après la perfusion.
Lorsque
plusieurs poches de Libmeldy sont nécessaires, il convient de s'assurer
avant la perfusion que le volume de médicament à administrer est
compatible avec la limite recommandée de DMSO, à savoir que le volume
total de DMSO administré doit rester inférieur à 1 % du volume
plasmatique estimé du patient. Le volume maximal de Libmeldy à
administrer doit donc rester inférieur à 20 % du volume plasmatique
estimé du patient (voir rubrique Précautions particulières d'élimination et de manipulation).
En outre, quand plus d'une poche de Libmeldy est nécessaire, il convient de perfuser une seule poche de médicament par heure.
Échec de prise de greffe
Au cours des études cliniques, aucun patient n'a subi d'échec de greffe de la moelle osseuse, tel que mesuré par la numération des neutrophiles dans le sang périphérique. L'échec de prise de greffe des neutrophiles est un risque à court terme qui peut potentiellement être grave, défini comme l'incapacité à atteindre un nombre absolu de neutrophiles (NAN) supérieur à 500 cellules/µL associée à l'absence de signes de rétablissement de la moelle osseuse (c'est-à-dire une moelle hypocellulaire) au jour 60 après la perfusion de Libmeldy. En cas d'échec de la prise de greffe, les cellules souches de secours non-transduites doivent être administrées par perfusion conformément aux normes locales (voir rubrique Posologie et mode d'administration).
Cytopénie prolongée
Les
patients peuvent présenter des cytopénies sévères, y compris une
neutropénie sévère [définie par une numération absolue des neutrophiles
(NAN) inférieure à500/µL] et une thrombocytopénie prolongée, pendant
plusieurs semaines après un conditionnement myéloablatif et une
perfusion de Libmeldy. Dans les études cliniques, la récupération
hématologique après traitement par le busulfan a été généralement
observée quatre à cinq semaines après la perfusion de Libmeldy. Dans
l'étude clinique avec la formulation cryoconservée (commerciale), la
prise de greffe des neutrophiles est survenue après une médiane (min,
max) de 36,5 jours (31 à 40 jours) après la thérapie génique. Les
patients doivent donc être surveillés pour détecter tout signe ou
symptôme de cytopénie pendant au moins 6 semaines après la perfusion.
Les globules rouges doivent être surveillés selon l'évaluation
médicale, jusqu'à la prise de greffe de ces cellules et la
récupération. Une transfusion de soutien de globules rouges et de
plaquettes doit être effectuée selon le jugement médical et les
pratiques de l'établissement. La détermination du nombre de cellules
sanguines et tout autre examen approprié doivent être rapidement
envisagés dès lors que des symptômes cliniques évoquant une anémie
apparaissent.
Si la cytopénie persiste au-delà de six à sept semaines malgré
l'utilisation de médicaments mobilisant les granulocytes, les cellules
souches de secours non transduites doivent être perfusées. Si la
cytopénie persiste malgré la perfusion de cellules souches de secours
non transduites, des traitements alternatifs doivent être envisagés.
Retard de la prise de greffe des plaquettes
La
prise de greffe des plaquettes est définie comme le premier de trois
jours consécutifs avec des valeurs plaquettaires ≥ 20 x 109/L
obtenues à différents jours après la perfusion de Libmeldy, sans
transfusion de plaquettes administrée pendant les sept jours précédant
immédiatement la période d'évaluation et durant celle-ci (jusqu'à 60
jours après la thérapie génique).
Au cours du développement clinique, 4/35 patients (11,4 %) ont signalé
un retard de prise de greffe des plaquettes (médiane : 73,5 jours,
intervalle de 65 à 109 jours) non corrélé à une augmentation de
l'incidence des saignements. Dans le cadre de la prise en charge
standard/prophylaxie standard, tous les patients faisant partie de
l'ensemble intégré pour les données de sécurité (N = 29) ont reçu un
support par transfusion de plaquettes. La numération plaquettaire doit
être surveillée conformément à l'évaluation médicale, jusqu'à la prise
de greffe de ces cellules et la récupération. Un support par
transfusion de plaquettes doit être apporté selon le jugement médical
et la pratique de l'établissement.
Acidose métabolique
Préalablement à un traitement par Libmeldy, la présence d'une acidose tubulaire rénale doit être évaluée en plus des risques liés au médicament de conditionnement et des risques liés à la procédure de thérapie génique, qui peuvent contribuer au développement d'une acidose métabolique. L'équilibre acido-basique doit être surveillé tout au long du conditionnement et jusqu'à ce que le patient ne soit plus soumis à un stress métabolique. Le médecin traitant doit envisager un traitement de remplacement par bicarbonate de sodium en même temps que tout autre traitement nécessaire, et viser à corriger tout effet(s) indésirable(s) concomitant pouvant contribuer à l'acidose métabolique.
Transmission d'un agent infectieux
Bien que Libmeldy soit soumis à des tests de stérilité et de recherche de mycoplasmes lors de la libération des lots, il existe un faible risque de transmission d'agents infectieux. Les professionnels de santé qui administrent Libmeldy doivent par conséquent surveiller les patients pour détecter tout signe ou symptôme d'infection après le traitement et les traiter de façon adéquate, si nécessaire.
Surveillance de la thyroïde
Des augmentations transitoires de la thyréostimuline (TSH), de la T4 libre (FT4 ; thyroxine) et de la T3 libre (FT3 ; tri-iodothyronine) ont été observées chez certains patients au cours des études cliniques. Étant donné que les troubles thyroïdiens pourraient potentiellement être masqués par une maladie grave ou être induits par une médication concomitante, la fonction et la structure de la thyroïde des patients doivent être évalués avant le traitement par Libmeldy. La fonction et la structure de la thyroïde doivent également être surveillées à court terme après le traitement, et plus tard si nécessaire.
Risque d'oncogenèse insertionnelle
Il existe un risque théorique de leucémie ou de lymphome après traitement par Libmeldy. En cas de détection de leucémie ou de lymphome chez un patient ayant reçu Libmeldy, des prélèvements sanguins doivent être effectués en vue d'une analyse du site d'intégration.
Anticorps anti-ARSA
Pendant
le développement clinique, des anticorps anti-ARSA (AAA) ont été
signalés chez cinq patients. Les titres étaient généralement faibles,
et tous les événements se sont résolus spontanément ou après un
traitement par rituximab (voir rubrique Effets indésirables). Aucun effet sur l'efficacité clinique ou la sécurité n'a été observé.
La surveillance des AAA est recommandée avant le traitement, entre 1 et
2 mois après la thérapie génique et ensuite à 6 mois, 1 an, 3 ans, 5
ans, 7 ans, 9 ans, 12 ans et 15 ans après traitement.
En cas de survenue de la maladie ou de progression importante de la
maladie, une surveillance supplémentaire des AAA est recommandée.
Tests sérologiques
Libmeldy n'a pas été étudié chez les patients infectés par le VIH-1, VIH-2, HTLV-1, HTLV-2, VHB, VHC ou les mycoplasmes.
Tous les patients doivent faire l'objet d'un dépistage pour le VIH-1/2,
HTLV-1/2, VHB, VHC et les mycoplasmes avant la mobilisation ou le
prélèvement de moelle osseuse afin de garantir l'acceptation de la
source cellulaire pour la fabrication de Libmeldy.
Utilisation des antirétroviraux
Les patients ne doivent pas prendre de médicaments antirétroviraux à compter d'au moins un mois avant la mobilisation et/ou le prélèvement de moelle osseuse et jusqu'à au moins 7 jours après la perfusion de Libmeldy (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Si un patient nécessite un traitement par antirétroviraux suite à une exposition au VIH/HTLV, l'instauration du traitement par Libmeldy doit être reportée jusqu'à ce qu'un western blot VIH/HTLV et un test de charge virale aient été effectués 6 mois après l'exposition.
Interférence avec les tests de dépistage du VIH
Les patients ayant reçu Libmeldy sont susceptibles de présenter un résultat positif aux tests d'amplification en chaîne par polymérase (PCR) pour le VIH en raison de l'insertion du provirus VLV, ce qui entraîne un résultat faux positif pour le VIH. Par conséquent, les patients qui ont reçu Libmeldy ne doivent pas faire l'objet d'un dépistage de l'infection par le VIH à l'aide d'un test PCR.
Don de sang, d'organes, de tissus et de cellules
Tout au long de leur vie, les patients traités par Libmeldy ne doivent faire aucun don du sang, d'organes, de tissus ou de cellules en vue d'une transplantation. Ces informations figurent sur la carte d'alerte patient qui doit être remise au patient après le traitement.
Après l'administration de Libmeldy
Après la perfusion, les procédures standard de prise en charge des patients après une greffe de CSPH doivent être respectées.
L'immunoglobuline G doit être maintenue au-dessus de 5 g/l pour
prévenir de possibles infections tardives (survenant plus de 100 jours
après le traitement) associées à une hypogammaglobinémie sévère,
résultant de l'aphérèse ou du prélèvement et du conditionnement de la
moelle osseuse.
Tout produit sanguin nécessaire dans les 3 mois suivant la perfusion de Libmeldy doit être irradié.
Teneur en sodium
Ce médicament contient 35 à 560 mg de sodium par dose, ce qui équivaut à 2 à 28 % de l'apport quotidien maximal recommandé par l'OMS de 2 g de sodium pour un adulte.
Résumé du profil de sécurité
La sécurité de Libmeldy a été évaluée chez 35 patients atteints de LDM.
La
durée médiane du suivi dans l'ensemble des données de sécurité
intégrées, incluant 29 patients traités par la formulation fraîche
(expérimentale), était de 4,51 ans (intervalle : 0,64 à 8,85 ans).
Trois patients sont décédés et un total de 26 patients est resté dans
la phase de suivi.
La durée médiane du suivi chez les 6 patients traités par la
formulation cryoconservée (commerciale) était de 0,87 an (intervalle :
0,0 à 1,47 an). Tous sont restés dans la phase de suivi (voir rubrique Propriétés pharmacodynamiques).
Étant donné la petite population de patients, les effets indésirables
présentés dans le tableau ci-dessous ne donnent pas une vue exhaustive
de la nature et de la fréquence de ces événements.
Le traitement par Libmeldy est précédé par des interventions médicales, à savoir la collecte de cellules souches hématopoïétiques par prélèvement de moelle osseuse ou mobilisation de sang périphérique avec du G-CSF, avec ou sans plérixafor, suivie d'une aphérèse, et le conditionnement myéloablatif (de préférence par busulfan), qui comportent leurs propres risques. Lors de l'évaluation de la sécurité d'un traitement par Libmeldy, il convient d'examiner le profil de sécurité et les informations approuvées sur les médicaments utilisés pour la mobilisation du sang périphérique et le conditionnement myéloablatif, en plus des risques liés à la thérapie génique.
Tableau récapitulatif des effets indésirables
Les événements indésirables sont listés ci-dessous par classe de système d'organes MedDRA et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10).
Table 2 Effets indésirables attribués à Libmeldy
| Classe de système d'organes | Très fréquent | Fréquent |
| Affections du système immunitaire | Test d'anticorps positif (anticorps anti-ARSA) | |
Table 3 Effets indésirables potentiellement attribués au conditionnement myéloablatif*
| Classe de système d'organes | Très fréquent | Fréquent |
| Infections et infestations | | Virémie à cytomégalovirus, pneumonie, infection à staphylocoque, infection urinaire, infection virale |
| Affections hématologiques et du système lymphatique | Neutropénie fébrile, neutropénie | Anémie, thrombocytopénie |
| Troubles du métabolisme et de la nutrition | Acidose métabolique | Surcharge liquidienne |
| Affections psychiatriques | | Insomnie |
| Affections du système nerveux | | Maux de tête |
| Affections respiratoires, thoraciques et médiastinales | | Épistaxis, douleur oropharyngée |
| Affections gastro-intestinales | Stomatite, vomissements | Ascite, diarrhée, Hémorragie gastro-intestinale, nausées |
| Affections hépatobiliaires | Hépatomégalie, Maladie veino- occlusive hépatique | Hypertransaminasémie |
| Affections de la peau et du tissu sous-cutané | | Exfoliation cutanée |
| Affections musculosquelettiques et du tissu conjonctif | | Douleur dorsale, douleur osseuse |
| Affections du rein et des voies urinaires | | Oligurie |
| Classe de système d'organes | Très fréquent | Fréquent |
| Affections des organes de reproduction et du sein | Insuffisance ovarienne | |
| Troubles généraux et anomalies au site d'administration | | Pyrexie |
| Investigations | | Augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase, test d'aspergillus positif |
* Sur la base de 29 patients ayant subi un conditionnement myéloablatif par busulfan dans l'ensemble intégré de données.
Description de certains effets indésirables
Présence d'anticorps contre l'ARSA
Cinq des 35 patients ont été testés positifs pour les anticorps
anti-ARSA (AAA) à divers moments après le traitement et ont présenté
l'événement « Test d'anticorps positif / Présence d'anticorps dirigés
contre l'arylsulfatase A » rapporté par l'investigateur.
Les titres d'anticorps étaient généralement faibles et se sont résolus,
soit spontanément, soit après un court traitement avec du rituximab.
Chez tous les patients ayant des résultats positifs au test AAA, aucun
effet négatif n'a été observé postérieurement au traitement au niveau
de l'activité ARSA dans les sous-populations cellulaires du sang
périphérique ou de la moelle osseuse, ni sur l'activité ARSA dans le
liquide céphalo-rachidien. Les patients traités par Libmeldy doivent
être surveillés régulièrement pour détecter la présence éventuelle
d'AAA (voir rubrique Mises en garde spéciales et précautions d'emploi).
Prélèvement de moelle osseuse et mobilisation de sang périphérique et aphérèse
Au cours des études cliniques, le profil de sécurité du prélèvement de
moelle osseuse et de la mobilisation/aphérèse était conforme à la
sécurité et à la tolérance connues des deux procédures et aux RCP des
agents de mobilisation (G-CSF et plérixafor).
Aucun événement indésirable grave n'a été signalé comme étant
potentiellement attribuable au prélèvement de moelle osseuse dans la
plage des volumes de moelle osseuse prélevés (le volume médian était de
35,5 mL/kg ; intervalle : 15,1 - 56,4 mL/kg). Dans l'ensemble intégré
des données de sécurité (n = 29), un patient a ressenti une douleur
osseuse, qualifiée d'événement indésirable de grade 2 et jugée liée à
la procédure de prélèvement de moelle osseuse, mais sans relation avec
le volume prélevé.
Aucun événement indésirable grave n'a été signalé comme potentiellement
attribuable à la mobilisation et à l'aphérèse, et aucun des patients
ayant subi une mobilisation n'a présenté d'événement indésirable au
cours de la phase de pré-traitement qui aurait pu être attribué aux
agents de mobilisation.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE AVANT le traitement
:
- Neurologique complet, fonction motrice et neurocognitive, selon l'âge du
patient.
- Fonction rénale.
- Fonction hépatique.
- Traitement
prophylactique de la maladie veino-occlusive et des complications liées aux
lésions endothéliales associées (microangiopathie thrombotique associée à une
greffe ) ou le syndrome hémolytique et urémique atypique .
- Traitement
prophylactique pour les convulsions et les infections (bactériennes, fongiques,
virales).
- Présence d'une acidose tubulaire rénale.
- Fonction et la
structure de la thyroïde .
- Anticorps anti-ARSA (AAA).
- Dépistage pour
le VIH-1/2, HTLV-1/2, VHB, VHC et les mycoplasmes avant la mobilisation ou le prélèvement de la moelle
osseuse.
- Numération plaquettaire
conformément à l'évaluation médicale, jusqu'à la prise de greffe de ces cellules
et la récupération.
SURVEILLANCE PENDANT le
traitement :
- Infections et événements liés au cathéter.
- Des patients qui n'avaient pas été exposés auparavant
au DSMO.
- Signes vitaux (pression artérielle, rythme cardiaque et saturation en oxygène) et apparition de tout symptôme d'hypersensibilité, avant le début de la perfusion, environ toutes les dix minutes pendant la perfusion, puis toutes les heures, pendant 3 heures, après la perfusion.
- L'équilibre acido-basique
tout au long du conditionnement et jusqu'à ce que le patient ne soit plus soumis
à un stress métabolique. Le médecin traitant doit envisager un traitement de
remplacement par bicarbonate de sodium en même temps que tout autre traitement
nécessaire, et viser à corriger tout effet(s) indésirable(s) concomitant pouvant
contribuer à l'acidose métabolique.
SURVEILLANCE APRES le traitement
:
- Signe ou symptôme de cytopénie pendant au moins 6 semaines après la
perfusion. Les globules rouges doivent être
surveillés selon l'évaluation médicale, jusqu'à la prise de greffe de ces
cellules et la récupération. Si la cytopénie
persiste au-delà de six à sept semaines malgré l'utilisation de médicaments
mobilisant les granulocytes, les cellules souches de secours non transduites
doivent être perfusées. Si la cytopénie persiste malgré la perfusion de cellules
souches de secours non transduites, des traitements alternatifs doivent être
envisagés.
- Signe ou symptôme d'infection.
- Fonction et la
structure de la thyroïde, à court terme après le traitement, et plus tard si
nécessaire.
- Anticorps anti-ARSA (AAA), entre 1 et 2 mois après la thérapie
génique et ensuite à 6 mois, 1 an, 3 ans, 5 ans, 7 ans, 9 ans, 12 ans et 15 ans
après traitement.
- L'immunoglobuline G doit être maintenue au-dessus de 5
g/l pour prévenir de possibles infections tardives (survenant plus de 100 jours
après le traitement) associées à une hypogammaglobinémie sévère, résultant de
l'aphérèse ou du prélèvement et du conditionnement de la moelle osseuse.
Tout
produit sanguin nécessaire dans les 3 mois suivant la perfusion de ce médicament
doit être irradié.
Interférence avec les tests de dépistage du
VIH
Les patients ayant reçu ce traitement sont susceptibles de
présenter un résultat positif aux tests d'amplification en chaîne par polymérase
(PCR) pour le VIH en raison de l'insertion du provirus VLV, ce qui entraîne un
résultat faux positif pour le VIH. Par conséquent, les patients qui ont reçu ce
traitement ne doivent pas faire l'objet d'un dépistage de l'infection par le VIH
à l'aide d'un test PCR.
Le médecin traitant doit informer les parents/soignants du patient des options de cryoconservation des cellules souches spermatogoniales ou du tissu ovarien.
Comme
Libmeldy n'est pas destiné à être utilisé chez les adultes, il n'existe
pas de données humaines sur l'utilisation pendant la grossesse ou
l'allaitement ni d'études sur la reproduction animale.
Concernant la fertilité, consultez le résumé des caractéristiques du
médicament de conditionnement myéloablatif. Il est à noter que le
médecin traitant doit informer les parents/soignants du patient des
options de cryoconservation des cellules souches spermatogoniales ou du
tissu ovarien.
La nature de Libmeldy est telle qu'aucune interaction pharmacocinétique n'est attendue avec d'autres médicaments.
Les patients ne doivent pas prendre de médicaments antirétroviraux à compter d'au moins un mois avant la mobilisation et/ou le prélèvement de moelle osseuse et jusqu'à au moins 7 jours après la perfusion de Libmeldy (voir rubrique Mises en garde spéciales et précautions d'emploi).
Vaccins vivants
La sécurité de l'immunisation par les vaccins viraux vivants pendant ou après le traitement par Libmeldy n'a pas été étudiée. La vaccination avec des vaccins à virus vivants n'est pas recommandée durant les 6 semaines précédant le début du conditionnement myéloablatif, et jusqu'à ce que le rétablissement hématologique soit effectué après un traitement par Libmeldy.
Libmeldy doit être administré dans un centre de traitement qualifié expérimenté dans la greffe de cellules souches hématopoïétiques (GCSH).
Les patients doivent être inclus et être suivis dans une étude de suivi à long terme, afin de mieux comprendre la sécurité et l'efficacité à long terme de Libmeldy.
Posologie
La dose de Libmeldy à administrer est définie en fonction du poids du patient au moment de la perfusion.
La dose minimale recommandée de Libmeldy est de 3 x 106 cellules CD34+ par kg. Dans les études cliniques, des doses allant jusqu'à 30 x 106 cellules CD34+ par kg ont été administrées.
Le volume maximal de Libmeldy à administrer doit être inférieur à 20 % du volume plasmatique estimé du patient (voir rubriques Mises en garde spéciales et précautions d'emploi et Précautions particulières d'élimination et de manipulation).
Libmeldy est destiné à un usage autologue (voir rubrique Mises en garde spéciales et précautions d'emploi) et ne doit être administré qu'une seule fois.
Prélèvement de moelle osseuse ou mobilisation de sang périphérique et aphérèse
Les cellules autologues de CD34+
sont isolées à partir d'un prélèvement de moelle osseuse (MO) ou de
sang périphérique mobilisé (SPm). Dans le cas où les cellules CD34+ sont isolées du SPm, une ou plusieurs procédures d'aphérèse seront effectuées après mobilisation du sang périphérique.
La décision d'utiliser la MO ou le SPm comme source pour l'isolement des cellules CD34+
est laissée à la discrétion du médecin traitant, en tenant compte de
l'âge et du poids du patient, de son état clinique et des conditions de
l'accès veineux.
En général, le SPm est la source cellulaire préconisée pour la
production de Libmeldy étant donné qu'elle est moins invasive pour le
patient.
La MO constituerait néanmoins la source cellulaire de choix chez les
nourrissons et les enfants pesant moins de 7 kg en cas de
contre-indication à l'utilisation de facteurs de croissance/agents de
mobilisation et lorsque l'accès veineux est considéré comme inadapté
pour la mise en place du cathéter d'aphérèse.
En fonction de la source cellulaire, le patient doit être en mesure de donner un minimum de 8-10 × 106 CD34+ cellules/kg, nécessaires à la production de Libmeldy (voir tableau 1).
Si les cellules CD34+ sont isolées à partir de la MO, lorsque cela est possible, la quantité minimale de cellules CD34+
doit être collectée en une seule procédure de prélèvement de MO. Avant
cette procédure, une première ponction de moelle osseuse est
généralement utilisée pour effectuer une numération initiale des
cellules, ce qui permet d'estimer le volume total de MO qui sera
nécessaire pour obtenir un nombre suffisant de cellules pour la
fabrication du médicament (voir rubrique Propriétés pharmacodynamiques).
Si les cellules CD34+ sont isolées du SPm, la quantité minimale de cellules CD34+ peut être atteinte en utilisant un ou plusieurs cycles d'aphérèse.
| Tableau 1 | Quantité de cellules CD34+ nécessaire à la production de Libmeldy en fonction de la source cellulaire (nombre de cellules exprimé en 106 cellules CD34+/kg) |
| Source cellulaire | Nombre minimum | Intervalle optimal |
| MO | 10 | 20-40 |
| SPm | 8 | 20-30 |
Si, après la fabrication du médicament, la dose minimale de Libmeldy de 3 × 106 cellules CD34+/kg n'est pas atteinte, le patient peut subir un nouveau prélèvement de moelle osseuse ou un nouveau protocole de mobilisation avec un ou plusieurs cycles d'aphérèse, afin d'obtenir plus de cellules pour une fabrication supplémentaire (voir Mobilisation et aphérèse à la rubrique Propriétés pharmacodynamiques).
Un prélèvement de secours de CSPH contenant au moins 2 x 106 cellules CD34+/kg
est également nécessaire pour être utilisé comme traitement de
remplacement si la qualité de Libmeldy est compromise après le début du
conditionnement myéloablatif et avant la perfusion de Libmeldy, en cas
d'échec de la greffe primaire ou d'aplasie médullaire prolongée après
le traitement par Libmeldy (voir rubrique Mises en garde spéciales et précautions d'emploi).
Ces cellules doivent être prélevées chez le patient au moment du
prélèvement de MO ou de l'aphérèse du SPm et doivent être
cryoconservées selon des procédures institutionnelles préalablement au
conditionnement myéloablatif.
Mobilisation du sang périphérique
Lorsqu'il est décidé d'utiliser le SPm comme source cellulaire, les
patients doivent subir une mobilisation des CSPH avec du facteur de
stimulation des colonies de granulocytes (G-CSF) avec ou sans
plérixafor, puis une aphérèse pour obtenir des cellules souches CD34+ en vue de la fabrication du médicament (voir rubrique Propriétés pharmacodynamiques pour une description du régime de mobilisation utilisé dans les études cliniques).
Conditionnement recommandé avant le traitement
Le médecin traitant doit confirmer que l'administration d'une thérapie
à base de CSPH autologues est cliniquement appropriée pour le patient
avant le début du conditionnement myéloablatif (voir rubrique Mises en garde spéciales et précautions d'emploi).
Un
conditionnement myéloablatif est nécessaire avant la perfusion de
Libmeldy afin de favoriser une prise de greffe efficace des cellules
CD34+ autologues génétiquement modifiées (voir rubrique Propriétés pharmacodynamiques pour une description du schéma myéloablatif utilisé lors des études cliniques).
Le busulfan est le médicament de conditionnement recommandé.
Le conditionnement myéloablatif ne doit pas commencer avant que le jeu
complet de poche(s) de perfusion constituant la dose de Libmeldy ait
été reçu et stocké au centre de traitement qualifié, et que la
disponibilité du prélèvement de secours soit confirmée.
Parallèlement au traitement de conditionnement, et avant le traitement
par Libmeldy, il est recommandé que les patients reçoivent un
traitement pour la prophylaxie de la maladie veino-occlusive (MVO) et
des complications liées aux lésions endothéliales associées,
c'est-à-dire la microangiopathie thrombotique associée à une greffe
(MAT-AG) ou le syndrome hémolytique et urémique atypique (SHUa),
conformément aux recommendations locales.
En fonction du schéma de conditionnement myéloablatif administré, une
prophylaxie pour les convulsions doit également être envisagée. La
phénytoïne n'est pas recommandée, car elle peut augmenter la clairance
du busulfan.
Une utilisation prophylactique et empirique des anti-infectieux
(bactériens, fongiques, viraux) doit être envisagée pour la prévention
et la prise en charge des infections, en particulier pendant la période
neutropénique consécutive au conditionnement. Une surveillance de
routine des virus les plus couramment sujets à réactivation est
recommandée conformément aux recommandations locales. Des mesures de
lutte contre les infections et des procédures d'isolement doivent être
employées pendant l'hospitalisation, conformément aux normes locales.
Prémédication
Il est recommandé d'administrer une prémédication avec de la
chlorphéniramine par voie intraveineuse (0,25 mg/kg, dose maximale de
10 mg), ou un médicament équivalent, 15 à 30 minutes avant la perfusion
de Libmeldy, afin de réduire le risque de réaction allergique à la
perfusion.
Populations particulières
Patients âgés
Libmeldy n'a pas été étudié chez les patients âgés de plus de 65 ans.
Insuffisance rénale
Libmeldy n'a pas été étudié chez les patients souffrant d'une
insuffisance rénale. Les patients doivent être évalués pour rechercher
une insuffisance rénale afin de s'assurer que l'administration d'une
thérapie génique autologue à base de CSPH est appropriée. Aucun
ajustement de la dose n'est nécessaire.
Insuffisance hépatique
Libmeldy n'a pas été étudié chez les patients présentant une
insuffisance hépatique. Les patients doivent être évalués pour
rechercher une insuffisance hépatique afin de s'assurer que
l'administration d'une thérapie génique autologue à base de CSPH est
appropriée. Aucun ajustement de la dose n'est nécessaire.
Population pédiatrique
La sécurité et l'efficacité de Libmeldy n'ont pas encore été établies
chez les patients atteints de la forme juvénile tardive de la maladie
(qui se déclare généralement après l'âge de 7 ans). Aucune donnée n'est
disponible.
Mode d'administration
Libmeldy doit être uniquement administré par perfusion intraveineuse (voir rubrique Précautions particulières d'élimination et de manipulation pour les détails complets sur la procédure d'administration).
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient des cellules humaines génétiquement modifiées.
Les professionnels de santé doivent par conséquent prendre les
précautions appropriées (porter des gants et des lunettes) pour éviter
toute transmission potentielle de maladies infectieuses lors de la
manipulation du produit.
Pour les instructions concernant la préparation, l'exposition accidentelle et l'élimination de Libmeldy, voir la rubrique Précautions particulières d'élimination et de manipulation.
Préparation pour perfusion
Avant la perfusion de Libmeldy, il doit être confirmé que l'identité du
patient correspond aux informations personnelles uniques du patient
figurant sur les étiquettes de la/des poches de perfusion et sur la
fiche d'information du lot qui l'accompagne.
La décongélation et la perfusion de Libmeldy doivent être coordonnées.
L'heure de début de la perfusion doit être confirmée à l'avance et la
décongélation doit être ajustée de sorte que Libmeldy soit disponible
pour la perfusion lorsque le patient est prêt. Afin de conserver la
viabilité du produit, il est recommandé d'administrer Libmeldy
immédiatement dès la fin de la décongélation.
L'administration doit être terminée dans les deux heures suivant la décongélation.
Administration
Administrer le produit en perfusion intraveineuse à l'aide d'un
cathéter veineux central. Lorsque plusieurs poches de Libmeldy sont
nécessaires, une seule poche de médicament doit être administrée par
heure. Chaque poche doit être administrée à un débit de perfusion qui
ne dépasse pas 5 mL/kg/h, en moins de 30 minutes environ. Le dispositif
d'administration recommandé est composé d'un kit de transfusion
sanguine équipé d'un filtre de 200 µm (voir rubrique Précautions particulières d'élimination et de manipulation).
Durée de conservation :
6 mois.
Une fois décongelé : maximum 2 heures à température ambiante (entre 20 °C et 25 °C).
Précautions particulières de conservation :
Les poches de perfusion de Libmeldy doivent être conservées dans la phase vapeur de l'azote liquide (< - 130 °C) jusqu'à leur décongélation et leur administration.
Conserver la (les) poche(s) de perfusion dans la (les) cassette(s) métallique(s). Ne pas recongeler après décongélation.
Pour les conditions de conservation du médicament après décongélation, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Aucune donnée clinique n'est disponible concernant le surdosage de Libmeldy.
Groupe pharmacothérapeutique : Autres agents hématologiques, code ATC : A16AB21.
Mécanisme d'action
Libmeldy est
une thérapie génique ex vivo à base de cellules souches et progénitrices
hématopoïétiques (CSPH) CD34+ autologues génétiquement modifiées. Les CSPH
CD34+ autologues sont prélevées dans la moelle osseuse (MO) ou le sang
périphérique mobilisé (SPm) du patient et transduites avec un vecteur
lentiviral (VLV ARSA), qui insère une ou plusieurs copies de l'acide désoxyribonucléique
complémentaire (ADNc) de l'ARSA humain dans le génome de la cellule de sorte
que les cellules génétiquement modifiées deviennent capables d'exprimer
l'enzyme ARSA fonctionnelle. Lorsqu'elles sont administrées au patient suite à
l'administration d'un régime de conditionnement myéloablatif, les cellules
génétiquement modifiées se greffent et sont capables de reconstituer le
compartiment hématopoïétique. Une sous-population des CSPH perfusées et/ou de
leur progéniture myéloïde est capable de migrer à travers la barrière
hémato-encéphalique vers le cerveau et de se greffer en tant que microglies
résidentes du système nerveux central (SNC) et macrophages périvasculaires du
SNC ainsi que macrophages endoneuraux du système nerveux périphérique (SNP).
Ces cellules génétiquement modifiées peuvent produire et sécréter l'enzyme ARSA
fonctionnelle, qui peut être absorbée par les cellules avoisinantes, selon un
processus connu sous le nom de « correction croisée », et utilisée pour
détruire ou empêcher l'accumulation de sulfatides nocifs.
Suite à une prise de greffe stable et réussie chez le patient, il est attendu
que les effets du médicament soient persistants.
Effets pharmacodynamiques
Une prise de greffe périphérique durable et stable de cellules génétiquement modifiées a été observée à partir d'un mois après l'administration de Libmeldy chez tous les patients évaluables. Un nombre de copies du vecteur (« vector copy number », VCN) persistant a également été observé dans les cellules CD34+ isolées de la moelle osseuse tout au long de la période de suivi. Ces résultats biologiques montrent une prise prolongée en plusieurs lignées de cellules corrigées par des gènes, ce qui est essentiel pour soutenir la production à long terme de l'ARSA et le bénéfice clinique à long terme qui en résulte.
À l'année 1
après traitement, la proportion de colonies dérivées de la MO abritant le
génome du VLV (% LV+) dans la population globale traitée était de 54,8 %
(intervalle : 20,0 % à 100 %, [N = 23]).
Lors de l'année 5, la proportion de colonies dérivées de la MO abritant le
génome du VLV (% LV+), était de 45,0 % (intervalle : 18,8 % à 90,6 % [n = 6, 4
formes infantiles tardives/Late infantile (LI) et 2 dans la forme juvénile
précoce/Early Juvenile (EJ)], indiquant une prise de greffe stable au fil du
temps dans la population traitée.
Une reconstitution de l'activité de l'ARSA dans le système hématopoïétique a été observée chez tous les patients atteints de LDM traités, avec une reconstitution progressive des taux d'ARSA dans les cellules mononucléées du sang périphérique (Peripheral Blood Mononuclear Cells, PBMC) qui ont atteint des valeurs comprises dans la plage de référence normale dans les 3 mois suivant le traitement et sont restées dans cette plage normale pendant toute la durée du suivi (voir figure 1).
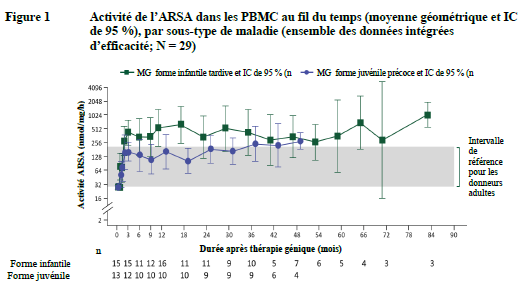
Remarque: les valeurs < LIQ sont attribuées à la LIQ. La LIQ est de 25,79 nmol/mg/h. Les MG et les IC à 95 % sont présentés lorsqu' il existe au moins 3 patients pour lesquels il n'y a pas de données manquantes. ARSA : arylsulfatase A ; IC : intervalle de confiance ; MG : moyenne géométrique ; LIQ : limite inférieure de quantification ; PBMC : cellules mononucléées du sang périphérique (peripheral blood mononuclear cells).
L'activité de l'ARSA a également été mesurée dans le liquide céphalo-rachidien (LCR) comme compartiment de substitution de la correction métabolique dans le cerveau. L'activité de l'ARSA dans le LCR est passée d'indétectable au moment de l'inclusion à détectable chez tous les patients évaluables dans le courant du 6e mois après traitement et a atteint des niveaux de référence à l'année 1 après traitement. Par la suite, la reconstitution centrale de l'activité enzymatique de l'ARSA est restée stable dans l'intervalle de référence.
Efficacité clinique
L'efficacité
clinique était basée sur l'analyse globale des résultats obtenus chez 29
patients atteints de la forme précoce de la LDM, traités par Libmeldy préparé
sous forme d'une formulation fraîche (non cryoconservée). Ces résultats ont été
obtenus chez vingt (20) patients traités dans le cadre de l'étude
d'enregistrement (étude 201222 - essai clinique ouvert, non randomisé, de
sécurité et d'efficacité à un seul bras) avec une durée médiane de suivi après
traitement de 4,0 ans (intervalle : 0,6 à 7,5 ans) et neuf (9) patients traités
dans le cadre de 3 programmes d'accès élargis avec un suivi médian de 1,5 ans
(intervalle : 0,99 à 2,72 ans).
De plus, les résultats initiaux obtenus auprès de 9 patients traités dans le
cadre d'une autre étude avec la formulation commerciale (cryoconservée) de
Libmeldy (étude 205756) sont résumés ci-dessous.
Le spectre de la LDM peut se présenter sous diverses formes cliniques, essentiellement en fonction de l'âge lors de l'apparition des premiers symptômes de la maladie. Les patients présymptomatiques atteints d'une forme infantile tardive (LI) ou d'une forme juvénile précoce (EJ) de LDM et les patients EJ présentant des symptômes précoces de LDM avec mutations bialléliques dans le gène de l'ARSA responsables d'une diminution de l'activité enzymatique de l'ARSA ont été inclus dans le développement clinique du Libmeldy. Les « mutations bialléliques dans le gène de l'ARSA responsables d'une diminution de l'activité enzymatique de l'ARSA » sont des mutations responsables d'un arrêt partiel ou complet de l'activité enzymatique de l'ARSA entraînant une accumulation des sulfatides. Ces mutations bialléliques excluent les mutations neutres communes décrites en association avec les allèles d'ARSA de pseudo-déficience.
Caractéristiques des patients et de la maladie
Les formes (variantes) de LDM ont été définies par la présence des critères suivants au cours du développement clinique :
· forme
infantile tardive (LI) : âge à l'apparition des symptômes chez le ou les frères
et sœurs plus âgés inférieur ou égal à 30 mois et/ou 2 allèles ARSA mutants
nuls (0) et/ou neuropathie périphérique à l'étude d'électroneurographie (ENG);
· forme
juvénile précoce (EJ) : âge à l'apparition des symptômes (chez le patient ou
chez le frère ou la sœur plus âgé[e]) entre 30 mois et avant 7 ans, et/ou un
allèle ARSA mutant nul (0) et un allèle ARSA mutant résiduel (R) et/ou
neuropathie périphérique à l'étude ENG.
Dans
la définition ci-dessus, les allèles nuls (0) ou résiduels (R) font référence à
des mutations connues ou nouvelles.
Le statut symptomatique des patients était défini comme suit:
· Pré-symptomatique : au moment de leur inclusion dans les études cliniques, les patients LI ou EJ ne présentaient pas d'insuffisance neurologique (symptômes liés à la maladie), avec ou sans signes de la maladie révélés par des évaluations instrumentales, c'est-à-dire l'étude électroneurographique (ENG) et l'imagerie par résonance magnétique (IRM) du cerveau.
Sur la base d'une analyse des caractéristiques initiales des patients LI et EJ pré-symptomatiques traités pendant le programme de développement clinique, la définition du statut pré-symptomatique a été affinée afin d'optimiser le bénéfice du traitement.
Compte tenu des résultats de cette analyse, le traitement par Libmeldy d'un patient pré-symptomatique doit être envisagé :
- Pour
les patients présentant la forme infantile tardive de la maladie, en l'absence
de retard dans la mise en place de la station debout indépendante ou de retard
dans l'obtention de la marche indépendante, associés à des signes anormaux lors
de l'évaluation neurologique.
- Pour
un patient atteint de la forme juvénile précoce de la maladie, en l'absence de
signes ou symptômes neurologiques de la maladie entraînant une altération ou
une régression des fonctions cognitives, motrices ou comportementales
(corroborée par un examen neurologique, une évaluation de la fonction motrice
brute et/ou des tests neuropsychologiques adaptés à l'âge).
Présence de
symptômes précoces : au moment de l'inclusion dans les études cliniques, les
patients EJ présentant des symptômes précoces répondaient aux 2 critères
suivants : quotient intellectuel (QI) ≥ 70 et
capacité à marcher de manière autonome en faisant ≥ 10 pas.
Sur la base de
l'analyse des bénéfices cliniquement pertinents sur les fonctions motrices et
cognitives, l'efficacité n'a été démontrée que chez les patients traités avant
l'apparition de la détérioration cognitive au moment où ils étaient encore
capables de marcher de manière indépendante.
Compte tenu de
ces résultats, le traitement par Libmeldy d'un patient souffrant d'une forme EJ
avec symptômes précoces de la maladie doit être envisagé :
- Si
ce patient est capable de marcher de manière indépendante, ce qui signifie que
son score GMFC- MLD (Gross Motor Function Classification dans la LDM) est ≤
1, et
- Si
la fonction cognitive du patient n'a pas commencé à décliner, ce qui signifie
que le patient a un QI ≥ 85.
Au moment de leur inclusion dans les études cliniques, sur les 29 patients atteints de LDM à apparition précoce, 20 étaient pré-symptomatiques et 9 présentaient des symptômes précoces, 16 présentaient un diagnostic de LDM LI et 13 présentaient un diagnostic de LDM EJ. Tous les patients LI de l'étude et certains patients EJ ont été identifiés après qu'un frère ou une sœur plus âgé(e) a développé des symptômes et reçu un diagnostic de LDM, ce qui a donné lieu à des tests chez d'autres membres de la famille.
Tableau 4 Résumé des caractéristiques démographiques par état symptomatique au moment de la thérapie génique et par sous-type de maladie (ensemble des données d'efficacité intégrée)
| Patients pré-symptomatiques | Patients présentant des symptômes précoces | |||
| Sous-groupe ayant une forme infantile tardive (N = 15) | Sous-groupe ayant une forme juvénile précoce (N = 5) | Sous-groupe ayant une forme infantile tardive (N = 1) | Sous-groupe ayant une forme juvénile précoce (N = 8) | |
| Sexe, n (%) | ||||
| Féminin | 5 (33) | 2 (40) | 1 (100) | 5 (63) |
| Masculin | 10 (67) | 3 (60) | 0 | 3 (38) |
| Âge au moment de la thérapie génique, en mois | ||||
| Médiane | 13,1 | 48,9 | 23,3 | 77,9 |
| Min | 7,6 | 11,4 | 23,3 | 38,8 |
| Max | 17,8 | 66,8 | 23,3 | 139,9 |
Prélèvement de moelle osseuse
Pendant le développement clinique, le volume de MO prélevé a été ajusté pour chaque patient. Le volume médian de MO recueilli était de 35 mL/kg (intervalle de 15 à 56 mL/kg), sans aucun événement associé en matière de sécurité.
Mobilisation et aphérèse
Au cours du développement clinique, tous les (dix) patients pour lesquels la décision a été prise d'utiliser le SPm comme source des cellules ont reçu du G-CSF (10-12,5 µg/kg/jour) pour mobiliser les cellules CD34+ avant la procédure d'aphérèse. À partir du 3e jour d'administration de G-CSF, un agent de mobilisation supplémentaire, le plérixafor, a été administré une fois par jour (0,24 mg/kg, par voie sous-cutanée), si cela était cliniquement indiqué en fonction de la numération des globules blancs et du nombre de cellules CD34+ dans le sang périphérique du patient. L'aphérèse a été effectuée dès que le nombre de cellules CD34+ a atteint un niveau adéquat, selon les procédures standard.
Si le nombre cible de cellules CD34+ collectées pour fabriquer Libmeldy et pour fournir les cellules du prélèvement de secours n'était pas atteint avec une seule aphérèse, une seconde procédure était effectuée. Pour tous les patients, le nombre minimum de cellules CD34+ nécessaire pour produire Libmeldy (8 x 106 cellules CD34+/kg) a été recueilli avec un cycle de mobilisation et 1 ou 2 aphérèses.
Conditionnement avant traitement
Tous les patients ont reçu un conditionnement systémique avec du busulfan avant le traitement par Libmeldy.
Treize patients (45 %) ont été traités par un régime de conditionnement submyéloablatif (SMAC), défini comme une ASC cumulée cible de 67,200 µg * h/l. Seize patients (55 %) ont été traités au moyen d'un régime de conditionnement myéloablatif (MAC), défini par une ASC cumulée cible de 85,000 µg * h/l.
Pour le schéma de conditionnement SMAC, les patients ont reçu au total 14 doses de busulfan (en fonction du poids du patient), en perfusion intraveineuse de 2 heures administrée toutes les 6 heures, du jour - 4 au jour - 1. Les taux plasmatiques de busulfan ont été surveillés par un prélèvement pharmacocinétique sanguin et ajustés à l'aide d'une ASC cible de 4800 µg*h/l (intervalle : 4200 à 5600 µg*h/l), ce qui correspond à une ASC cumulée totale attendue de 67,200 µg*h/l (intervalle : 58,800 à 78,400 µg*h/l). L'ASC cumulée moyenne chez les patients ayant reçu un schéma SMAC était plus élevée que prévue, mais restait dans la plage cible (moyenne géométrique 71,923.53 [IC à 95 % : 68751,04, à 75242,41]).
Pour le schéma de conditionnement MAC, les patients ont reçu une dose de busulfan basée sur la surface corporelle en fonction de leur âge (80 mg/m2/dose si ≤ 1 an; 120 mg/m2/dose si > 1 an) pour un total de 4 doses, administrées par perfusion intraveineuse de 3 heures toutes les 20 à 24 heures entre le jour - 4 et le jour - 1. Les taux plasmatiques de busulfan ont été surveillés au moyen d'un prélèvement pharmacocinétique en série et ajustées en utilisant une ASC totale cumulée cible de 85 000 µg*h/l (intervalle : 76 500 - 93 500 µg*h/l).
Les analyses de sous-groupes par schéma de conditionnement, c'est-à-dire la comparaison des sous-groupes de patients ayant reçu le schéma MAC et de ceux ayant reçu le schéma SMAC, n'ont pas montré de différences notables dans le niveau de prise de greffe des cellules transduites ni dans l'activité enzymatique de l'ARSA (dans les PBMC totales et les cellules mononucléées dérivées de la MO). En outre, les profils de sécurité des deux schémas se sont avérés comparables.
Par conséquent, la décision d'utiliser le régime MAC ou SMAC pour le conditionnement préalable au traitement est laissée à la discrétion du médecin traitant, compte tenu des caractéristiques cliniques du patient telles que, sans s'y limiter, l'âge, la fonction hépatique, la prématurité et la thrombophilie.
Pendant le développement clinique, une prophylaxie de la maladie veino-occlusive (MVO) et des complications liées à une lésion endothéliale était requise selon la pratique de l'établissement avec l'acide ursodésoxycholique ou le défibrotide.
Administration de Libmeldy
Tous les patients (N = 29) ont reçu le médicament avec une dose moyenne (min, max) de 10,81 x 106 (4,2 ; 25,9) cellules CD34+/kg administrée en perfusion intraveineuse.
Résultats d'efficacité intégrés (N=29)
Les co-critères principaux d'évaluation de l'efficacité étaient les suivants:
· Mesure
de la fonction motrice globale (Gross Motor Function Measure, GMFM) : Une
amélioration de > 10 % du score GMFM total chez les patients traités par
rapport aux scores GMFM dans une population de patients LDM du même âge non
traités correspondant à un contrôle historique (c'est-à- dire l'étude TIGET sur
l'histoire naturelle de la maladie [HN]), évaluée à l'Année 2 après le
traitement (voir Tableau 5), et
· Activité de ARSA : Une augmentation significative (≥ 2 ET) de l'activité de l'ARSA résiduelle par rapport aux valeurs avant traitement, mesurée dans les cellules mononuclées du sang périphériques (PBMC) 2 ans après le traitement (voir Effets pharmacodynamiques, figure 1 et tableau 6).
Les patients
atteints de LDM à apparition précoce et traités avant l'apparition des
symptômes manifestes ont montré un développement moteur normal, une
stabilisation ou un retard dans la vitesse de progression du dysfonctionnement
moteur, mesuré par le score total GMFM (%) (voir tableau 5). Selon un modèle
ANCOVA ajusté pour l'âge lors de l'évaluation de la GMFM et du traitement, la
différence moyenne entre les patients LI pré-symptomatiques traités et les
patients LI du même âge non traités provenant de l'étude HN était de 71,0 % à
l'année 2 et de 79,8 % à l'année 3. De même, la différence moyenne entre les
patients EJ pré-symptomatiques traités et les patients EJ du même âge non traités
était de 52,4 % à l'année 2 et de 74,9 % à l'année 3. Ces différences de
traitement étaient statistiquement significatives (p ≤ 0,008) en faveur
de Libmeldy.
Bien qu'elle
ne soit pas statistiquement significative, une différence nette dans le score
total GMFM a également été notée entre les patients EJ présentant des symptômes
précoces traités et les patients EJ non traités et appariés selon l'âge (28,7 %
à l'année 2 ; p = 0,350 et 43,9 % à l'année 3 ; p = 0,054).
Tableau 5 Score total de la GMFM (%) à l'année 2 et l'année 3 chez les patients pré-symptomatiques et symptomatiques précoces (sous-groupes avec la forme infantile tardive et juvénile précoce) en comparaison avec les données d'histoire naturelle dans une population du même âge (ensemble des données d'efficacité intégrée).
| Score total GMFM moyen ajusté | Différence moyenne de traitement quant au score GMFM total entre les patients traités et les patients de l'histoire naturelle non traités appariés en fonction de l'âge | |||
| Patients traités | Patients de l'histoire naturelle non traités | |||
| Patients pré- symptomatiques | Forme infantile tardive | |||
| Année 2 * | 79,5 % (n = 10) | 8,4% (n=8) | 71,0% (95% IC: 60,4 - 81,7) ; p<0.001 | |
| Année 3 | 82,6% (n=9) | 2,8% (n=9) | 79,8% (95% IC: 66,2 - 93,3); p = 0,001). | |
| Forme Juvénile précoce | ||||
| Année 2 * | 96,7% (n=4) | 44,3% (n=8) | 52,4% (95% IC: 25,1 - 79,6) ; p=0.008 | |
| Année 3 | 93,2% (n=4) | 18,2% (n=9) | 74,9% (95% IC: 50,8 - 99,1) ; p<0.001 | |
| Patients symptomatiques précoces |
Forme Juvénile précoce
|
|||
| Année 2 * | 60,7% (n=6) | 31,9% (n=10) | 28,7% (95% IC: -14,1 - 71,5) ; p=0.350 | |
| Année 3 | 59,8% (n=6) | 15,9% (n=10) | 43,9% (95% IC: 9,2 - 78,5) ; p=0,054 | |
* La mesure de la fonction motrice globale deux ans après le traitement constituait un co-critère principal d'évaluation de l'étude clinique d'enregistrement. Remarque: analyse de l'ajustement de la covariance en fonction du traitement et de l'âge. Les valeurs p proviennent d'un test d'hypothèse à 5 % à deux bornes avec hypothèse nulle d'une différence de 10 %. IC : intervalle de confiance ; EJ : juvénile précoce (early juvenile) ; GMFM : mesure de la fonction motrice motrice globale (gross motor function measurement) ; LI : infantile tardive (late infantile) ; LDM : leucodystrophie métachromatique.
La détérioration de la fonction motrice globale a été évaluée à partir de l'apparition de la maladie chez les patients EJ qui présentaient des symptômes précoces au moment de la thérapie génique. Quatre ans après l'apparition de la maladie, la proportion estimée de patients ayant survécu et conservé leur locomotion et leur capacité à s'asseoir sans soutien (niveau 5 ou supérieur de GMFC-LDM) était de 62,5 % dans le groupe traité, contre 26,3 % dans le groupe non traité, ce qui représente un retard de progression de la maladie après un traitement par Libmeldy.
Une augmentation statistiquement significative de l'activité ARSA dans les PBMC a également été observée 2 ans après la fin du traitement, par comparaison à la référence avant traitement, chez les patients pré-symptomatiques (20,0 fois plus ; p < 0,0001) et chez les patients à symptômes précoces (4,2 fois plus ; p = 0,004) (Voir tableau 6).
Tableau 6 Activité de l'ARSA, mesurée dans les PBMC (moyenne géométrique) au départ et 2 ans après traitement chez des patients pré-symptomatiques et les patients à symptômes précoces (ensemble des données d'efficacité intégrée).
| Moyenne géométrique (coefficient de variabilité interindividuelle, CV%)) Activité de l'ARSA dans les PBMC | Facteur de multiplication entre la référence et l'Année 2* | ||
| Valeur initiale | Année 2: | ||
| Pré- | 26,923 (6,72) | 339,736 (270,85) | 20,0 |
| symptomatique | (n=19) | (n=14) | (95% IC: 9,0 - 44,0) p < 0,001 |
| Symptomatique | 26,025 (2,72) | 134,056 (55,94) | 4,2 |
| précoce | (n=9) | (n=6) | (95% CI: 1,6 - 11,2) p = 0,004 |
* Ratio en moyennes ajustées à partir d'un modèle mixte pour mesures répétées des données sur l'échelle logarithmique, en ajustant pour la visite, référence, référence*visite, sous-type de maladie et sous-type de maladie*visite
Un critère
d'évaluation d'efficacité secondaire de l'analyse d'efficacité intégrée était
la mesure d'un QI supérieur à 55 après le traitement par Libmeldy, le seuil de
retard mental modéré (DSM-IV), à l'aide de tests neuropsychologiques. Les
mesures du quotient intellectuel/quotient de développement (QI/QD),
c'est-à-dire les capacités cognitives et linguistiques, complètent les
résultats du score GMFM et fournissent des preuves supplémentaires que les
niveaux élevés de prise de greffe et de reconstitution enzymatique se
traduisent par des effets thérapeutiques pertinents sur les principaux domaines
symptomatiques des patients atteints de LDM.
Dans le
sous-groupe LI (tous les patients pré-symptomatiques au moment du traitement
sauf un), 12 patients sur 15 évalués présentaient un QI/QD assez constant, dans
l'intervalle normal (score QI/QD de 100 ± ET de 15) tout au long du suivi. Tous
ces patients sauf 2 (un pré-symptomatique, un à symptômes précoces) sont restés
au-dessus du seuil de handicap mental sévère (QI/QD > 55) à des âges chronologiques
auxquels les 14 patients HN non traités ayant subi des évaluations
neuropsychologiques ont montré des signes de handicap cognitif sévère
(c'est-à-dire QI/QD inférieur à 55 et proche de 0).
Sur les 10
patients EJ qui ont survécu, les 4 patients pré-symptomatiques et 4 patients
sur 6 présentant des symptômes précoces ont montré des valeurs normales du
QI/QD tout au long du suivi. En revanche, au cours du suivi, 11 des 12 patients
HN ayant fait l'objet d'évaluations neuropsychologiques ont présenté des signes
de dysfonctionnement cognitif sévère.
Au moment de
l'analyse des données intégrées, c'est-à-dire à une période de suivi médiane de
3,035 ans après le traitement (intervalle de 0,99 à 7,51 ans), aucun des 16
patients du sous-groupe LI traité, tous pré-symptomatiques au moment du
traitement sauf un, n'était décédé (survie globale de 100 %). Quatre patients
LI pré-symptomatiques étaient vivants 6 ans ou plus après le traitement et 2
patients LI pré-symptomatiques étaient en vie 7 ans ou plus après le traitement.
En comparaison, 12 patients LI sur 19 (63,2 %) non traités dans l'étude HN
étaient décédés au moment de l'analyse.
Une survie
globale comparable a été observée dans les groupes EJ traités ou non traités
avec une durée de suivi médiane de 3,49 ans après traitement (intervalle de
0,64 à 6,55 ans). Un patient EJ sur 5 (20 %) traité
à un stade pré-symptomatique est décédé à la suite d'un infarctus ischémique
cérébral, qui n'a pas été considéré comme étant lié à Libmeldy. Deux décès ont
été enregistrés parmi les 8 (25,0 %)
patients EJ traités au stade symptomatique précoce, les deux en raison d'une
progression de la maladie. Eux non plus n'ont pas été considérés comme étant
liés au traitement par Libmeldy. De même, 3 des 12 (25 %) patients EJ non
traités dans l'étude HN étaient décédés au moment de l'analyse.
Une analyse de
sensibilité réalisée pour identifier les facteurs cliniques qui auraient pu
influencer le niveau de bénéfice du traitement avec Libmeldy et optimiser les
recommandations d'utilisation du traitement, a identifié 4 échecs du traitement
:
- Un
patient LI a présenté des symptômes liés à la maladie entre l'étape de
sélection et l'administration de Libmeldy et a été considéré comme
symptomatique au moment du traitement. La progression de ce patient après
traitement était comparable à celle des patients HN non traités tant au niveau
de la fonction cognitive que du développement moteur.
- Trois
patients EJ symptomatiques précoces traités par Libmeldy ont présenté une
détérioration des fonctions motrices et cognitives comparable à celle observée
chez des patients HN non traités, et la progression de la maladie a entraîné le
décès de deux d'entre eux. Deux des trois patients présentaient un QI inférieur
à 85 (82 et 58) au moment du traitement. Deux des trois patients ont connu une
détérioration entre les évaluations de sélection et les évaluations de
référence (début du schéma de conditionnement).
Étude 205756 (formulation commerciale cryoconservée)
L'étude 205756 est une étude ouverte à un seul bras visant à évaluer la formulation cryoconservée (commerciale) de Libmeldy dans le traitement des patients atteints de LDM LI pré-symptomatiques et de LDM EJ pré-symptomatiques et à symptomes précoces. La gamme de doses de cellules utilisée chez les 9 premiers patients de l'étude 205756 (10,45-30,0 x 106 cellules CD34+/kg) est proche de celle utilisée chez les patients traités avec la formulation fraîche (expérimentale) du médicament (4,2- 25,9 x 106 cellules CD34+).
Au moment de la date de fin de recueil des données, 6 patients (3LIs, 3EJs), tous pré-symptomatiques au moment du traitement, ont été traités, avec un suivi médian post-traitement de 0,87 an (intervalle : 0,0 à 1,47 an). Les données préliminaires d'efficacité montrent des niveaux de prise de greffe, de nombre de copies du vecteur, d'activité de l'ARSA dans les PBMC et le LCR à différents moments après la thérapie génique, qui sont de l'ordre de ceux observés dans l'analyse intégrée des données des patients traités avec la formulation fraîche de Libmeldy.
Les données préliminaires relatives à la sécurité indiquent que Libmeldy a été bien toléré. Le profil de sécurité observé dans cette étude avec la formulation cryoconservée est conforme au profil établi chez les patients traités avec la formulation fraîche en termes de nature, de délai d'apparition et de fréquence des effets indésirables signalés.
Population pédiatrique
Libmeldy a été étudié chez des nourrissons et des enfants âgés de 7,6 mois à 11,6 ans. L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Libmeldy dans le sous-groupe de patients « juvénile tardif » (late juvenile) de la population pédiatrique atteinte de leucodystrophie métachromatique (c'est-à-dire les patients atteints de LDM âgés de 7 ans à moins de 17 ans au moment de l'apparition de la maladie) (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Libmeldy est un médicament de thérapie génique composé de cellules autologues qui ont été génétiquement modifiées ex vivo. La nature de Libmeldy est telle que les études conventionnelles de pharmacocinétique, d'absorption, de métabolisme et d'élimination ne sont pas applicables. La biodistribution de Libmeldy a néanmoins été étudiée et sa diffusion dans les tissus hématopoïétiques et les organes cibles de la maladie (y compris le cerveau) a été démontrée.
Libmeldy n'a aucun effet sur l'aptitude à conduire des véhicules et à utiliser des machines. L'effet des agents de mobilisation et de l'agent de conditionnement myéloablatif sur l'aptitude à conduire des véhicules ou à utiliser des machines doit être pris en compte.
En raison de la nature de Libmeldy, une évaluation toxicologique standard n’était pas applicable et les
études conventionnelles de mutagénicité, de carcinogénicité et de toxicité pour la reproduction et le
développement n’ont pas été menées.
La pharmacologie, la toxicologie et la génotoxicité de Libmeldy ont été évaluées in vitro et in vivo.
L’analyse du site d’intégration (integration site analysis, ISA) des cellules Lin- issues de la moelle osseuse de souris et des cellules CD34+ humaines transduites avec le VLV ARSA a été réalisée avant et après la greffe chez la souris et n’a montré aucun enrichissement de l’insertion dans les gènes liés au cancer ou à proximité de ceux-ci, ni de dominance clonale. Un prototype de vecteur lentiviral correspondant au VLV ARSA n’a pas induit de transformation in vitro ni de croissance durable des cellules Lin- issues de la moelle osseuse de souris de type sauvage en lien avec transformation insertionelle. Les cellules Lin- issues de la moelle osseuse de souris Cdkn2a-/-, une souche sujette au cancer déclenché par la mutagenèse insertionnelle gamma-rétrovirale, transduites avec le même prototype de vecteur lentiviral, n’ont pas montré de potentiel génotoxique lorsqu’elles ont été transplantées sur des souris de type sauvage.
Des études de toxicité et d’oncogenèse (tumorigénicité) ont été réalisées dans le modèle murin de la LDM. Aucun signe de toxicité due à une surexpression de l’ARSA et aucune croissance anormale ou maligne de cellules transplantées ou de tumeurs hématopoïétiques liées à l’intégration du VLV ARSA n’ont été observés. La surexpression de l’ARSA dans les CSPH humaines et chez les souris ARSA génétiquement modifiées n’a pas altéré l’activation d’autres sulfatases dépendantes du facteur activant les sulfatases SUMF-1, n’a pas affecté les capacités de prolifération et de différenciation des cellules transduites et n’a pas induit de toxicité ou de déficience fonctionnelle dans l’ARSA des souris génétiquement modifiées.
Des études supplémentaires avec des cellules CD34+ humaines transduites avec le VLV ARSA administré à des souris immunodéficientes et ayant subi une myéloablation n’ont montré aucune toxicité, aucune mobilisation du vecteur et aucune transduction de proximité (bystander) des gonades mâles.
La surveillance moléculaire n’a pas permis de détecter de réplication lentivirus compétente (replication competent lentivirus, RCL).
La pharmacologie, la toxicologie et la génotoxicité de Libmeldy ont été évaluées in vitro et in vivo.
L’analyse du site d’intégration (integration site analysis, ISA) des cellules Lin- issues de la moelle osseuse de souris et des cellules CD34+ humaines transduites avec le VLV ARSA a été réalisée avant et après la greffe chez la souris et n’a montré aucun enrichissement de l’insertion dans les gènes liés au cancer ou à proximité de ceux-ci, ni de dominance clonale. Un prototype de vecteur lentiviral correspondant au VLV ARSA n’a pas induit de transformation in vitro ni de croissance durable des cellules Lin- issues de la moelle osseuse de souris de type sauvage en lien avec transformation insertionelle. Les cellules Lin- issues de la moelle osseuse de souris Cdkn2a-/-, une souche sujette au cancer déclenché par la mutagenèse insertionnelle gamma-rétrovirale, transduites avec le même prototype de vecteur lentiviral, n’ont pas montré de potentiel génotoxique lorsqu’elles ont été transplantées sur des souris de type sauvage.
Des études de toxicité et d’oncogenèse (tumorigénicité) ont été réalisées dans le modèle murin de la LDM. Aucun signe de toxicité due à une surexpression de l’ARSA et aucune croissance anormale ou maligne de cellules transplantées ou de tumeurs hématopoïétiques liées à l’intégration du VLV ARSA n’ont été observés. La surexpression de l’ARSA dans les CSPH humaines et chez les souris ARSA génétiquement modifiées n’a pas altéré l’activation d’autres sulfatases dépendantes du facteur activant les sulfatases SUMF-1, n’a pas affecté les capacités de prolifération et de différenciation des cellules transduites et n’a pas induit de toxicité ou de déficience fonctionnelle dans l’ARSA des souris génétiquement modifiées.
Des études supplémentaires avec des cellules CD34+ humaines transduites avec le VLV ARSA administré à des souris immunodéficientes et ayant subi une myéloablation n’ont montré aucune toxicité, aucune mobilisation du vecteur et aucune transduction de proximité (bystander) des gonades mâles.
La surveillance moléculaire n’a pas permis de détecter de réplication lentivirus compétente (replication competent lentivirus, RCL).
Précautions à prendre avant la manipulation ou l'administration du médicament
· Ce médicament contient des cellules
sanguines humaines génétiquement modifiées. Les professionnels de santé qui
manipulent Libmeldy doivent prendre les précautions appropriées (port de gants,
de vêtements de protection et de lunettes de protection) pour éviter toute
transmission potentielle de maladies infectieuses.
· Libmeldy doit se trouver à tout moment à une température
inférieure à - 130 °C, jusqu'à ce que le contenu de la poche soit décongelé
pour perfusion.
Définition de la dose à administrer
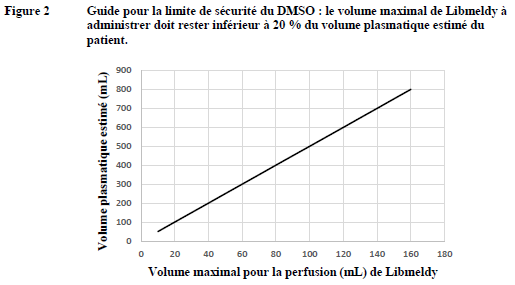
· Compte tenu des informations posologiques fournies à la rubrique Posologie et mode d'administration, la dose à perfuser et le nombre de poches de perfusion à utiliser doivent être définis en fonction du nombre total de cellules CD34+ fournies indiqué sur la fiche d'information du lot (c'est-à-dire la « dose fournie » calculée en fonction du poids du patient au moment de la collecte des cellules). La dose de Libmeldy à administrer doit également tenir compte du poids du patient au moment du traitement et du fait que toute poche utilisée doit être administrée dans son intégralité.
· Le volume de perfusion
doit faire l'objet d'une attention particulière en fonction de l'âge et du
poids du patient. Lorsque la dose de Libmeldy à perfuser représente plus d'une
poche, il convient de s'assurer avant la perfusion que le volume de médicament
à administrer est compatible avec la limite recommandée de DMSO, c'est-à-dire
que le volume total de DMSO administré doit rester
< 1 % du volume plasmatique estimé du patient. Par conséquent, le volume
maximal de Libmeldy à administrer doit rester inférieur à 20 % du volume
plasmatique estimé du patient.
· Le graphique suivant est fourni à titre de référence afin de déterminer le volume maximal de Libmeldy qui peut être administré par perfusion à un patient en fonction de son volume plasmatique estimé.
Préparation de la perfusion
· Il est possible qu'un
patient nécessite plusieurs poches de perfusion. Chaque poche de perfusion est
fournie dans un sac de suremballage, contenu dans une cassette métallique.
· La ou les poches de
perfusion emballées dans les sacs de suremballage doivent être conservées à
l'intérieur de leurs cassettes métalliques dans la phase de vapeur de l'azote
liquide à < - 130 °C, jusqu'à ce qu'elles soient prêtes à être décongelées
et perfusées.
· Prendre en compte
toutes les poches de perfusion et confirmer pour chaque poche de perfusion
qu'elle n'a pas dépassé la date de péremption à l'aide de la fiche
d'information du lot jointe.
· Une solution stérile de
chlorure de sodium à 9 mg/mL (0,9 %) pour injection doit être disponible pour
amorcer la tubulure avant la perfusion et pour rincer la poche de perfusion et
la tubulure après la perfusion.
Inspection avant décongélation
· Ne pas retirer la
cassette métallique du stockage cryogénique ou décongeler Libmeldy avant que le
patient ne soit prêt à recevoir la perfusion. Le moment de la décongélation de
la ou des poches de perfusion contenant Libmeldy et celui de la perfusion
doivent être coordonnés. Confirmer l'heure de perfusion à l'avance et ajuster
l'heure de début de décongélation afin que le traitement soit disponible pour
la perfusion lorsque le patient est prêt.
· Ouvrir la cassette
métallique et vérifier l'intégrité du sac de suremballage et de la poche de
perfusion avant la décongélation. Si une poche de perfusion est abîmée, suivre
les recommandations locales pour la manipulation des déchets de matières
d'origine humaine et contactez Orchard Therapeutics immédiatement.
· Avant de décongeler
Libmeldy, il faut vérifier que l'identité du patient correspond aux
informations uniques sur le patient indiquées sur les étiquettes des emballages
et sur la fiche d'information du lot qui les accompagne. Libmeldy est réservé
seulement à un usage autologue. Ne pas décongeler ou perfuser Libmeldy si les
informations figurant sur l'étiquette spécifique au patient sur la poche de
perfusion ne correspondent pas au patient en question.
Décongélation
· Après avoir
soigneusement retiré la poche de perfusion de la cassette métallique,
décongeler celle- ci dans son sac de suremballage scellé à 37 °C dans un
dispositif de décongélation contrôlée jusqu'à ce qu'il n'y ait plus de glace
visible dans la poche de perfusion.
· Une fois la
décongélation terminée, le sac doit être immédiatement retiré du dispositif de
décongélation.
· Ouvrir soigneusement le
sac de suremballage pour retirer la poche de perfusion, qui doit être conservée
à température ambiante (entre 20 °C et 25 °C) jusqu'à la perfusion.
· Masser doucement la
poche de perfusion pour remettre les cellules en suspension. Le contenu de la
poche de perfusion doit être inspecté pour détecter tout reste visible
d'agrégat cellulaire. Les petits agrégats cellulaires doivent être dispersés en
les mélangeant délicatement manuellement. Ne pas secouer la poche.
· La poche de perfusion
ne doit pas être lavée, tournée vers le bas, faire l'objet d'un prélèvement
et/ou être remise en suspension dans un autre milieu avant la perfusion.
· Libmeldy ne doit pas
être irradié car l'irradiation pourrait entraîner une inactivation du produit.
· Si plusieurs poches de
perfusion sont prévues pour la dose de traitement du patient, la poche suivante
ne doit être décongelée qu'après perfusion complète du contenu de la poche
précédente.
Administration
· Libmeldy doit être administré sous forme de
perfusion intraveineuse par l'intermédiaire d'un cathéter veineux central,
conformément aux procédures habituelles du centre d'administration des produits
de thérapie cellulaire.
· Le dispositif d'administration recommandé est
composé d'un kit de transfusion sanguine équipé d'un filtre de 200 µm.
· Chaque poche doit être perfusée par gravité
dans les 2 heures suivant la décongélation, y compris toute interruption
pendant la perfusion, afin de préserver la viabilité maximale du produit.
· Le débit de perfusion maximal est de 5 mL/kg/h,
et le contenu de chaque poche doit être perfusé en 30 minutes environ.
· Lorsque plusieurs poches de Libmeldy sont
nécessaires, il convient de perfuser une seule poche de produit par heure.
· Les patients qui n'ont jamais été exposés
auparavant au DMSO doivent faire l'objet d'une surveillance étroite. Les signes
vitaux (tension artérielle, fréquence cardiaque et saturation en oxygène) et
l'apparition de tout symptôme doivent être surveillés pendant les 3 heures
suivant la perfusion.
· À la fin de la perfusion, rincer la totalité de
Libmeldy restant dans la poche de perfusion et toute tubulure associée avec une
solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) pour s'assurer que
le plus grand nombre possible de cellules soit administré au patient. Le volume
de perfusion doit faire l'objet d'une attention particulière en fonction de
l'âge et du poids du patient.
Précautions à prendre pour l'élimination du médicament
· Libmeldy contient des cellules humaines
génétiquement modifiées. Les procédures locales sur la manipulation de matériel
d'origine humaine concernant la gestion des médicaments non utilisés ou
l'élimination les déchets doivent être suivies.
· Tout matériel ayant été
en contact avec Libmeldy (déchets solides et liquides) doit être manipulé et
éliminé comme des déchets potentiellement infectieux, conformément aux
procédures locales sur la manipulation de matériel d'origine humaine.
Exposition accidentelle
· Il convient d'éviter toute exposition accidentelle à Libmeldy. Les procédures locales relatives à la manipulation de matériaux d'origine humaine doivent être suivies en cas d'exposition accidentelle, ce qui peut inclure le lavage de la peau contaminée et le retrait des vêtements contaminés. Les surfaces de travail et le matériel susceptibles d'avoir été en contact avec Libmeldy doivent être décontaminés avec un désinfectant approprié.
Liste I.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Prescription réservée aux spécialistes et services
NEUROLOGIE.
Prescription réservée aux spécialistes et services PEDIATRIE.
Réservé à l'usage HOSPITALIER.
Dispersion pour perfusion.
Dispersion transparente à légèrement trouble, incolore à jaune ou rose.
Poche(s) de perfusion de 50 mL en éthylène-acétate de vinyle (EVA) munie(s) de deux dispositifs de perfusion disponibles, chacune emballée dans un sac de suremballage en EVA et placée à l'intérieur d'une cassette métallique.
Libmeldy est expédié de l'usine de fabrication au lieu de stockage du centre de traitement dans un conteneur pour cryoconservation, qui peut contenir plusieurs cassettes métalliques destinées à un seul patient. Chaque cassette métallique contient une seule poche de perfusion de Libmeldy.
2.1 Description générale
Libmeldy (atidarsagène autotemcel) est une thérapie génique contenant une population autologue enrichie en cellules CD34+ qui contient des cellules souches et progénitrices hématopoïétiques (CSPH) transduites ex vivo avec un vecteur lentiviral codant le gène de l'arylsulfatase A (ARSA) humaine.
2.2 Composition qualitative et quantitative
Le médicament se compose d'une ou plusieurs poches de perfusion contenant une dispersion de 2-10 x 106 cellules/mL en suspension dans une solution cryoconservatrice. Chaque poche de perfusion contient 10 à 20 mL de Libmeldy.
Comme le nombre total de cellules et la concentration de cellules CD34+ varient selon les lots individuels des patients, les informations quantitatives concernant le dosage (concentration totale de cellules viables), le volume de dispersion et le nombre total de cellules CD34+ par poche et la dose du médicament sont indiquées dans la fiche d'information du lot. La fiche d'information du lot est incluse avec la boîte cryogénique utilisée pour transporter Libmeldy.
Excipients à effet notoire
Ce médicament
contient 3,5 mg de sodium par mL et 55 mg de diméthylsulfoxyde (DMSO) par mL.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Diméthylsulfoxyde
Chlorure de sodium
Albumine humaine